DISCONTINUATIONS
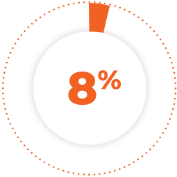
of patients permanently discontinued treatment due to an adverse reaction. The adverse reaction resulting in permanent discontinuation in ≥2% of patients was second primary malignancy.1
8% of patients permanently discontinued treatment due to an adverse reaction. The adverse reaction resulting in permanent discontinuation in ≥2% of patients was second primary malignancy.1
REDUCTIONS
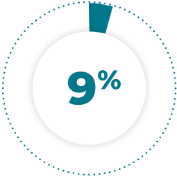
of patients receiving TAZVERIK® required dose reductions due to an adverse reaction.1
9% of patients receiving TAZVERIK® required dose reductions due to an adverse reaction.1
INTERRUPTIONS
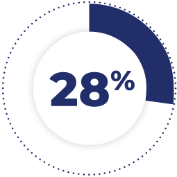
of patients receiving TAZVERIK required dose interruptions due to an adverse reaction. Adverse reactions requiring dosage interruptions in ≥3% of patients were thrombocytopenia and fatigue.1
28% of patients receiving TAZVERIK required dose interruptions due to an adverse reaction. Adverse reactions requiring dosage interruptions in ≥3% of patients were thrombocytopenia and fatigue.1
Serious adverse reactions occurred in 30% of patients who received TAZVERIK. Serious adverse reactions occurring in ≥2% of patients taking TAZVERIK included general physical health deterioration, abdominal pain, pneumonia, sepsis, and anemia.1
The most common (≥20%) adverse reactions were fatigue (36%), upper respiratory tract infection (30%), musculoskeletal pain (22%), nausea (24%), and abdominal pain (20%).1
Adverse reactions (≥10%) in patients with relapsed or refractory (R/R) FL who received TAZVERIK (N=99)1
ADVERSE REACTION |
ALL GRADES (%) |
GRADE 3 OR 4 (%) |
|---|---|---|
General |
||
|
Fatigue* |
36 |
5 |
|
Pyrexia |
10 |
0 |
Infections |
||
|
Upper respiratory tract infection† |
30 |
0 |
|
Lower respiratory tract infection‡ |
17 |
0 |
|
Urinary tract infection§ |
11 |
2 |
Gastrointestinal |
||
|
Nausea |
24 |
1 |
|
Abdominal pain‖ |
20 |
3 |
|
Diarrhea |
18 |
0 |
|
Vomiting |
12 |
1 |
Musculoskeletal and connective tissue |
||
|
Musculoskeletal pain¶ |
22 |
1 |
Skin and subcutaneous tissue |
||
|
Alopecia |
17 |
0 |
|
Rash# |
15 |
0 |
Respiratory and mediastinal system |
||
|
Cough** |
17 |
0 |
Nervous system |
||
|
Headache†† |
13 |
0 |
≤5% of patients experienced Grade 3 or 4 adverse reactions.1
Clinically relevant adverse reactions occurring in <10% of patients who received TAZVERIK included1:Infection: sepsis (2%), pneumonia (2%), and herpes zoster (2%)
- *Includes fatigue and asthenia.
- †Includes laryngitis, nasopharyngitis, pharyngitis, rhinitis, sinusitis, upper respiratory tract infection, viral upper respiratory tract infection.
- ‡Includes bronchitis, lower respiratory tract infection, tracheobronchitis.
- §Includes cystitis, urinary tract infection, urinary tract infection staphylococcal.
- ‖Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper.
- ¶Includes back pain, limb discomfort, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal pain, myalgia, neck pain, non-cardiac chest pain, pain in extremity, pain in jaw, spinal pain.
- #Includes erythema, rash, rash erythematous, rash generalized, rash maculopapular, rash pruritic, rash pustular, skin exfoliation.
- **Includes cough and productive cough.
- ††Includes headache, migraine, sinus headache.
R/R=relapsed or refractory.
Reference: 1. TAZVERIK (tazemetostat) Prescribing Information. Cambridge, MA: Epizyme, Inc., August 2024.
Select laboratory abnormalities (≥10%) worsening from baseline in patients with R/R FL who received TAZVERIK1
LABORATORY ABNORMALITY |
TAZVERIK* |
||
|---|---|---|---|
ALL GRADES (%) |
GRADE 3 OR 4 (%) |
||
Hematology |
|||
|
Decreased lymphocytes |
57 |
18 |
|
|
Decreased hemoglobin |
50 |
8 |
|
|
Decreased platelets |
50 |
7 |
|
|
Decreased white blood cells |
41 |
9 |
|
|
Decreased neutrophils |
20 |
7 |
|
Chemistry |
|||
|
Increased glucose |
53 |
10 |
|
|
Increased aspartate aminotransferase |
24 |
0 |
|
|
Increased alanine aminotransferase |
21 |
2.3 |
|
|
Increased alkaline phosphatase |
18 |
1.0 |
|
|
Increased creatinine |
17 |
0 |
|
*The denominator used to calculate the rate varied from 88 to 96 based on the number of patients with a baseline value and at least 1 posttreatment value.
TAZVERIK does not require special monitoring for laboratory abnormalities.1
R/R=relapsed or refractory; FL=follicular lymphoma.
Reference: 1. TAZVERIK (tazemetostat) Prescribing Information. Cambridge, MA: Epizyme, Inc., August 2024.